User login
News
Tue, 10/11/2022 - 13:59
Mon, 03/28/2022 - 14:40
Fri, 11/01/2019 - 20:13
Wed, 10/24/2018 - 21:14
Wed, 10/03/2018 - 14:48
TC_Comb
 TC_Comb: the wrapper of THERMOCALC program for effective multi-equilibrium geothermobarometry by the avPT method with visualization and analysis of results
TC_Comb: the wrapper of THERMOCALC program for effective multi-equilibrium geothermobarometry by the avPT method with visualization and analysis of results
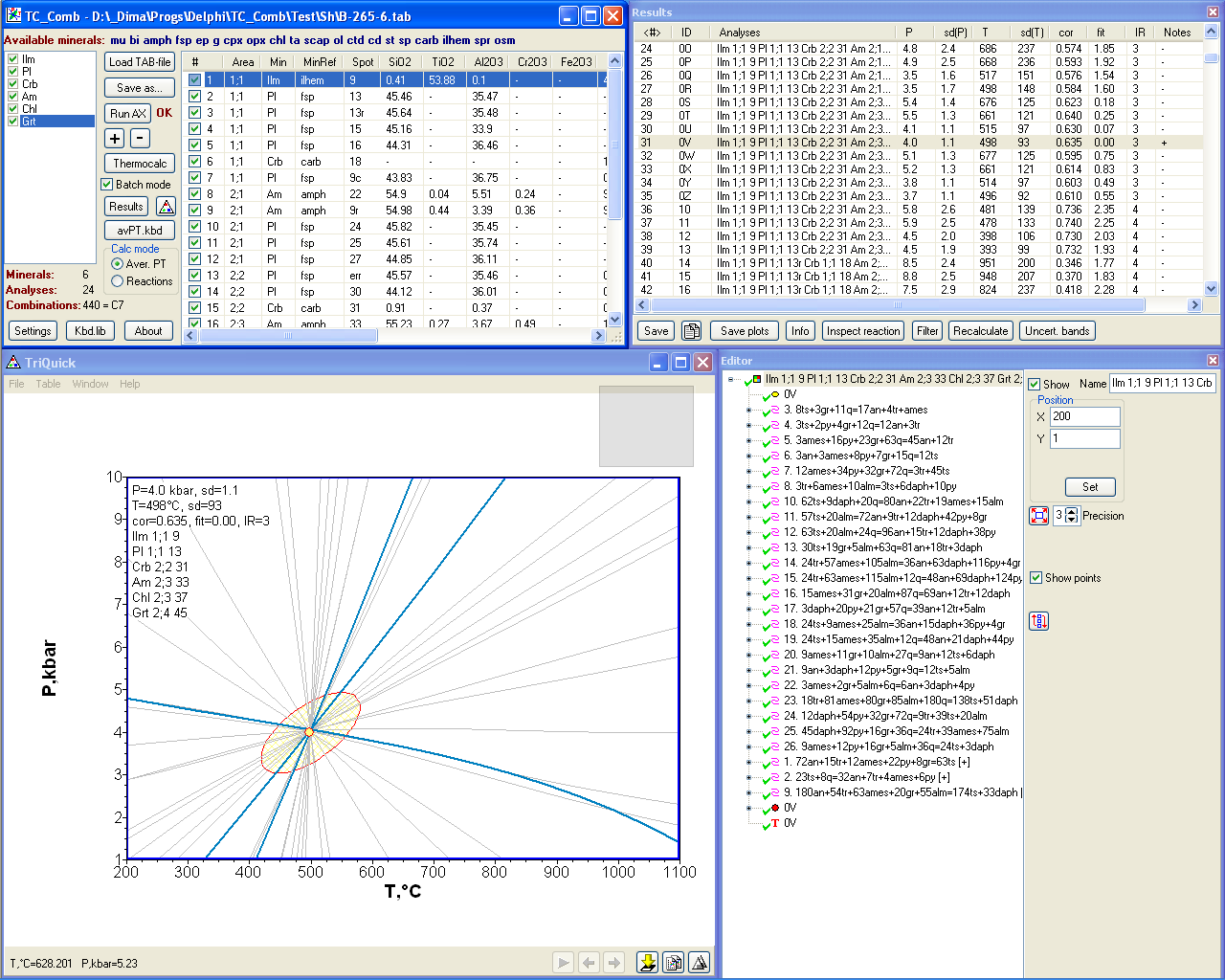
TC_Comb is a ©MS Windows wrapper for the THERMOCALC program of R. Powell and T. Holland running in multi-equilibrium geothermobarometry modes (only avPT+Reactions now). This program can make THERMOCALC geothermobarometry more effective and comfortable.
The main program features are:
- easy and fast entering of input data;
- fast (re)calculation of end-member activities using AX2 or AX62 programs of T. Holland;
- primitive but effective permutational approach for calculations of PT-parameters based on many sets of mineral analyses made from a single small area of thin section;
- estimation of H2O and CO2 activities or mole fractions (since version 1.1);
- additional functions: filtering of reactions by the presence of specified components, tracing of selected reaction line through a series of plots, etc.;
- visualization (plotting of PT-averages, error ellipses, reaction curves and their uncertainty bands) makes results of THERMOCALC thermobarometry more transparent for users. TriQuick (another program which acts as a server and editor of diagram graphics) is required for working with diagrams;
- output of all required information in tabular form.
TC_Comb is a freeware application.
NB! The last version of THERMOCALC which supports old (traditional) avPT mode is 3.45, newer versions are not compatible with TC_Comb!
Read about important changes in TC_Comb version 1.1
How to work with TC_Comb vers. 1.0 (basic operations demo):
If you like these programs and want to support their development, you can make a donation here
Comments
Hi,
Hi,
Thanks for creating this software, it looks very promising!
Here's a first question for you: we can't find anything in the help menu... We see the topics, but no text. Any idea?
Thanks!
Hello,
Hello,
This is a known issue with CHM help files related to evolution of Microsoft security politics (any downloaded CHM-files are regarded now as potentially dangerous objects and blocked by default). The solution is:
1. Right-click the .CHM file and choose "Properties"
2. On the "General" tab, click the button labeled "Unblock"
3. Click "OK"
It is from http://support.microsoft.com/kb/2021383.
Another source (with pictures and one more cause): http://weblog.helpware.net/?p=36
I'm also planning to duplicate documentation in PDF format.
I get this alert from TC-COMB
I get this alert from TC-COMB : Error double calculation at the same calcTatP varable detected (T/P) possibly due to inconsistent file Rct.kbd (use/create another one for this system). calculations terminated.
How can I solve this problem. I am not expert on thermocalc.
Hi,
Hi,
The simplest solution:
- place original file Kbd.lib from the distributive to TC_Comb directory (if this file has been modified while experimenting with interactive runs of Thermocalc in TC_Comb),
- press the "Kbd.lib" button in TC_Comb,
- select "Default" in the list (the first item),
- press the "Use" button (TC_Comb will show message about copying of avPT.kbd and Rct.kbd).
Some background info:
Thermocalc can calculate two kinds of tables with PT-coordinates of reaction curves: either T for the range of fixed P values (CalcTatP=Yes) or P for the range of fixed T values (CalcTatP=No). Default keyboard macro in TC_Comb (Rct.kbd) creates both of them one after another, then the program operates all the data simultaneously to avoid problems with sub-isobaric and sub-isothermal [parts of] curves.
While you can create/modify/select keyboard macros in TC_Comb, it's not necessary to use these features in regular work and the standard "Default" set of macros is enough. So, there is no need to use buttons "Kbd.lib" and "avPT.kbd" (or "Rct.kbd") and to uncheck "Batch mode" below the button "Thermocalc" (i.e. interactive runs of Thermocalc are not required usually in TC_Comb). I think I should add the option like "Expert mode" to the program to hide these buttons and checkbox...
Reply to the question
Reply to the question appeared on the TWQ_View page (http://www.dimadd.ru/en/comment/7#comment-7):
Hello,
It seems that this is the problem in TC_Comb, not TWQ_View...;)
Anyway, yes: this is due to cordierite! This mineral requires input of H2O and CO2 activities in Thermocalc even if the system is "dry" (doesn't contain explicit fluid components in the tc-ax.txt file). Therefore the keyboard macro "Default" is not suitable in this case and you should try another one. Press the "Kbd.lib" button in TC_Comb, select the macro named "Cordierite (aH2O & aCO2)" in the opened window and press the "Use" button.
BTW, you can double-click on any record in the "Results" window (even with failed calculations) and look at the Thermocalc screen output in the separate window. Type of calculations (avPT or Rct) can be selected in the main TC_Comb window. This way may be more convenient than checking of tc-log.txt.
Cheers,
Dmitry
Hi Dimitry,
Hi Dimitry,
First thank you very very much for your software that actually reconcile me with Thermocalc ;) So much easier to use it now, and the ability to plot reactions in just one click is just awesome! I do have a problem, however, for some calculation in upper amphibolite / lower granulite facies rocks. I am working with Al-rich pelite composed of quartz, plagioclase, K-feldspar, biotite, cordierite, and sillimanite. I successfully run AX and got the correct activity for all phases (and added as pure minerals quartz and sillimanite). Plagioclase activity has both an and ab components, and K-feldspar activity are "san" rich with some "ab" component. The problem is that I cannot use BOTH plagioclase AND K-feldspar in the same Thermocalc calculation. TC_Comb ask me to choose plagioclase OR K-feldspar, with the error message "Similar phases found: Pl and Kfs. Use Pl?"... Is this a bug or a limitation of the software?
Thanks in advance for your help!
Julien Allaz
Hi Julien,
Hi Julien,
Thank you for the high appreciation of the program! Yes, it is the sad THERMOCALC (and TC_Comb) limitation: we can not use several phases consisting of the same components in a single avPT session. Thus, for example, two-felspar thermometry is not available. However, it's possible to get around this problem.
First of all, this system (Crd+Bt+Pl+Kfs+Qtz+Sil) has only one Ca-bearing component "an": it means that no an-bearing reactions can be written, therefore "an" should be excluded. The same situation is with "ab": it is the only Na-bearing component of the system. So, plagioclase is a useless phase at all ;) and you can select Kfs only.
Though, "an" is important for garnet-bearing rocks (due to reactions with grossular). If "an" is a required component, you can select plagioclase for "an" activities and add "san" + its activity to the list of pure minerals together with quartz and sillimanite, i.e. something like this:
q sill san 0.93 H2O
You can try different activities (e.g. extremal "san" activities taken from analytical data) to see dependence of results on them.
NB! It's important that all reactions with "san" in these systems are associated with the decomposition of biotite (the second K-bearing phase), that is why I've added also H2O to the list of pure phases. Another important feature of this Grt-absent system: there are only 3 linearly independent reactions, or 2 IR if water is excluded :(
If cordierite and H2O are used, you should select the proper keyboard macro (use the "Kbd.lib" button to do this). "H2O and CO2 activities" is a good choise in this case. Then you can set some fixed aH2O and aCO2 values in the "Settings" window and perform calculations with them. After this you can select e.g. "Fixed: aCO2" (under the "aH2O" and "aCO2" input fields), check the "Calc fluid" checkbox instead of "Fluid from the table" (at the bottom of the "Results" window) and press the "Recalculate" button for selected successful combinations which demonstrate some PT results. TC_Comb will try to find the most suitable aH2O value for fixed aCO2. You can also try to select "Fixed: none" (to find aH2O and aCO2 simultaneously). However, I've found that THERMOCALC fails at some aCO2 values for this system... :(
Great suggestion! Thanks for
Great suggestion! Thanks for this quick and more than complete response.
Yes, the rock are Grt-bearing (an omission in my previous post). Typical new garnet growth after Bt-related melting (however, some biotite remain stable after this reaction). I already have beautiful PT-estimate using TWQ, and I was looking for a comparison of the absolute PT-data with Thermocalc. To my surprise, the temperature estimate with Thermocalc and only Pl OR Kfs yield slightly lower temperature than TWQ, but roughly the same (within the error). I will try the trick with san 0.93, and also make sure to include H2O (I can only assume pure water, although some CO2 might be present. I might try also an evaluation of the "optimum" H2O-CO2. My experience - 10 years ago! - with Thermocalc and amphibolite facies pelite yielded relatively coherent aH2O values, around 0.9, which is what I expected with Al-rich pelite. On the contrary, TWQ was often giving me abnormal results (0.3 xH2O or less).
You also have a good point about Na... I always keep it thinking about the paragonite component in muscovite and the albite, but of course muscovite is long gone in this sample, and therefore there is no more Na-exchange possible... Except maybe with cordierite, but there is very small amount of Na in these cordierite (around 0.1 wt-%) and I doubt there are any good thermodynamic data for Na-cordierite, yet.
Best,
Julien
A small remark about water:
A small remark about water: water activity may be substantially reduced not only because of addition of CO2 but also due to other fluid components (like NaCl, etc.) or due to melting. Thus, just H2O-CO2 binary is a too simple model and it's better to work only with activities of H2O and CO2 rather than with mole fractions. aCO2 in this system is required only for "tuning" of cordierite activity and its value may be too far from reality. Therefore "inconsistent" aH2O/aCO2 values are quite possible and should not be surprising.
Though, I have also obtained unrealistic aH2O values with TWQ (near zero!) but for "true" Hbl amphibolites (using database with amphiboles from Mader & Berman, 1992). According to our experience, TWQ with BA96A.DAT (we call it "granulitic database":) usually calculates reasonable water activities, especially if reactions in the "dry" subsystem are well converged.
Hi
Hi
I am working with TC_Comb and i want to calculate average PT for my analysis but i can not run the thermocalc section of this program, i am not expert in thermocalc. any body can help me?
Thanks
Hi Zahra,
Hi Zahra,
1. First of all, TC_Comb must know where the Thermocalc executable and desired database are installed. You can press the Settings button, select the TC and AX tab, then press buttons TC program and/or TC data base and choose required files (e.g. tc340.exe and tc-ds55.txt).
2. I've recognized that sometimes TC_Comb doesn't create default script files (tc-prefs.txt and tc-pt.txt) at its first run, this may cause problems. You can create these files manually (you can find their contents in the program manual), or look inside distributives on GoogleFrive and YandexDisk: I've just included these files there.
Anyways, read detailed description of avPT method on the Thermocalc website (archived). Also, the article Powell R., Holland T. (1994) Optimal geothermometry and geobarometry // American Mineralogist, v.79, p.120-133 may be very helpful.
HELLO
HELLO
Guys i need assistance on pasting my electron microprobe data on a software platform. I get the message saying ID number is absent.
Hello,
Hello,
Tables for TC_Comb should contain columns with mineral abbreviations and with IDs (numbers) of analyses. These identifiers are required to know which analysis of some mineral is used in particular combination. TC_Comb determines the column with IDs of analyses by its header. You can find list of possible headers in TC_Comb.ini, line #44:
Analysis=Point Spot Probe Analysis An
You can freely edit this list and add headers used in your own microprobe data tables, but make sure that it is not intersected with the other editable lists with headers (for columns with mineral abbreviations, area ID, totals and selection flags).
The program has been updated
The program has been updated to 1.1.0.5:
Hi Dmitry,
Hi Dmitry,
Thank you for making TC_Comb. I have been using it with Windows 10 for a couple of weeks and it's very helpful and robust.
I have a question about your calculation of error ellipses. In Holland and Powell (1988, page 179) they describe using a value for n of ~2.5 for an error ellipse of 95% confidence. However, it seems that you use ~1.25 in TC_Comb. Can you explain why you use 1.25? I know others do this too, but I don't see the reason.
Thanks,
Eric
Hi Eric,
Hi Eric,
Thank you for the interesting and important question!
It's not so easy to answer it in two words, but I will try...
First of all, I've implemented plotting of error ellipses in TriQuick, where ellipses may by described in two forms: pure geometric notation (center coordinates, width, height, slope) and statistical parameters (center coordinates, 2 sigmas on X and Y, correlation coefficient, variance, confidence level). TriQuick re-calculates statistical parameters to geometry before drawing. You can find and edit error ellipse statistical parameters in TriQuick (with a loaded TC_Comb diagram): open the "Editor" window, select error ellipse in the tree of objects and press the button "Stat.". TriQuick can also calculate error ellipses for clouds of points on XY scatter diagrams (plotted using its "Table" and "Variables" windows) for desired confidence levels. I've tested this functionality using independent statistical software (e.g. freeware PAST3) and I'm satisfied with it. I've placed the working algorithm to the Excel workbook ErrorEllipse.xls (you can download it from GoogleDrive or YandexDisk), it may be helpful.
The next difficult question was: what are these sd(T) and sd(P) values in the THERMOCALC output in fact? I've tried to reproduce error ellipse on Fig. 3 from Powell and Holland (1994, page 124) using data on activities from the Table 4 (the same paper, page 128), old THERMOCALC v.2.4 and old database TH.PD (generated 23.05.90), supposing the ellipse was calculated for conf. level 95%. Almost the same ellipse (but at somewhat lower PT) can be reproduced only if sd(T) and sd(P) are treated as 2 sigmas, not 1 sigma. Therefore I decided to place sd(T) and sd(P) values to TVL files "as is" without doubling them. I've put parameters calculated for this ellipse to the Excel workbook avept_DDD.xlsx (GoogleDrive,YandexDisk) as the first sample. This file is primarily the avept.xls of David Waters but with some my additions (just to compare results): Bi-scalar function approximation used in TriQuick (rough but enough precise for ellipses, I think) and drawing of better ellipse.
I'm not sure (as usual))) that this way is entirely correct but I hope that is so.
Doubt
When I put to generate the results, gives an error "Error calculating PT; Incomplete IR-set".
"THERMOCALC 3.33 (Free Pascal version)
the main output is in the file, "tc-PT-o.txt"
other (eg drawpd) output is in the file, "tc-PT-dr.txt"
calcs use:
reading a-x datafile, "tc-ax.txt"...
an ab tr fact ts parg
an ab tr fact ts parg
which end-members :
fact excluded
specification of PT window:
PT window: 5.0 <=> 25.0 kbar; 100 <=> 1500¡C (from script)
reactions :
no reactions
** no reactions
Average PT
BOMBED in posdefinvert : sqrt negative
hit return to exit program"
Do you have any idea why you are giving this error?
Doubt
Hi Luiz,
Your system contains only plagioclase (ab, an) and amphibole (tr, ts, parg, fact), so actually no stoichiometrically balanced reactions can be written between these very few components. Unfortunately, Thermocalc database does not contain edenite and richterite required for Hbl-Pl geothermometric equilibria proposed in Holland & Blundy (1994).
While one reaction can be written if quartz is added to the system (8Qtz + 2Parg = 2Ab + Tr + Ts), Thermocalc could not find it in some cases (because it goes far outside PT window).
TC_Comb under Windows 10 (64 bit)
The good news: TC_Comb is now compatible with MS Windows 10 (64 bit), the programs do not freeze after running THERMOCALC in batch mode.
A few words about technical details (for interested users):
Although THERMOCALC is an excellent program, it is not a perfectly programmed software: it contains bad errors, such us insufficient cheking for illegal operations. For example, if you specified a mineral assemblage with only two linearly independent reactions for avPT calculations, THERMOCALC doesn't check this, proceeds and crashes after division by zero (which is an OS level critical error). Thus, if 1000 combinations are planned for batch mode, and many of them crash THERMOCALC, you will have to manually close tons of OS notification windows like "The program performed illegal operation...". It's a good idea to handle THERMOCALC critical errors in TC_Comb (without passing them to the OS): this will prevent users from annoying OS notification windows. This is why TC_Comb launches THERMOCALC via a small debugger. This self-written debugger contained some bugs (and still contains, I'm pretty sure)), but it worked well until Windows 10 (64 bit), where a different DOS-emulator was implemented (all console applications work under the DOS emulator). This emulator creates new threads, but the previous version of the debugger routine did not care about this and froze all operations. The corrected debugger version follows created threads.
P.S. However, I noticed that batch THERMOCALC oprerations are processed 3-4 times slower in Windows 10 (64 bit) than in previous Windows versions.
TC_Comb simple question
Dear Dima,
I was trying to use your tool TC_Comb. Unfortunately, I had some difficulties with the calculation of end-members activities. I pasted required data to the main window of the program and saved it as a tab file. After that, I ran AX program I tried to open just saved tab file. However, I obtained an error message. I'm sure that I'm doing some simple mistake, but I don't know how to overcome this obstacle.
I would be grateful for any kind of advice
Jacek
Update. Finally, I have found the Comb.txt file that is placed in the folder with the AX program. Uff :-)
TC_Comb simple question
Dear Jacek,
I am very glad that you have overcome this problem on your own!:) Of course, AX program has its own specific format of input files, TC_Comb writes them separately to the AX directory. If you have several versions of AX program, it's also important to load Comb.txt in AX from the correct folder (i.e. from the folder of currently used AX version), sometimes Windows (or AX) "forget" to change folder after changing AX version.
TC_Comb simple question
Dear Dima,
I still have some difficulties with using TC Comb. At this point, I went through the calculation of and mebmber activities. However, there is no info concerning the activity of chl (chlorite). The system is composed of 7 minerals and 15 analyses. However, the major issue is related to further steps of the analysis. When I press the button Thermocalc only the first permutation is being calculated and I'm obtaining an error message that "calculation terminated (Timeout 7 sec at avpPT). Possible reasons: 1) infinite I/O loop due to inconsistent avpPT.kbd file, 2) Voluminous calculations)". I tried to look for this kbd file but it seems as it is lacking. I'm using TC version 3.5 and AX version 62.
I would like to kindly ask what could be the solution to this problem.
best regards
Jacek
TC_Comb simple question
Dear Jacek,
You've encountered two sad problems at once:
I have some collection of older TC and AX versions (+ databases), you can download this archive from GoogleDrive or YandexDisk.
TC_Comb simple question
Dear Dima,
thank you very much for your prompt reply and a link to all this stuff related to Thermocalc. It was very useful. Now it works much better (i.e. activities for chlorite end-members are now being calculated). However, I still have some strange difficulties. When I'm trying to work in a batch mode I obtain an error message that starts with statement that "calculations terminated ..." and so on. But when I'm working not in a batch mode I'm able to do the calculations (as I can see the results in terms of calculated P and T in a black Thermocalc window).
So, the question is what I'm doing wrong this time?
Now, I'm working using TC3.37 version, tc-ds55s.txt database and AX_2.exe.
best regards
Jacek
TC_Comb simple question
Dear Jacek,
This behaviour may point to inconsistent keyboard macros (stored in avPT.kbd, and Rct.kbd). The are many macros available via "Kbd.lib" button, selection of correct one depends on system composition: does it contain H2O, CO2 or cordierite, what variables for fluid components are to be used (activities or mole fractions)? If fluid components and cordierite absent, selection of "Default" macro should work well. Interactive mode (with batch mode unchecked) is to be used only for recording keyboard macros, it is generally not required at all because "Kbd.lib" already contains all possible variants of avPT session (I beleive so). In any case, use it carefully and don't save kbd file if you're not sure.
You can also double-click on any failed combination in the "Results" window, wait a little and check Thermocalc output. It may help to localize problem.
TC_Comb simple question
Dear Dima,
I'm still struggling with TC_Comb. Now, I'm able to do some calculations and a window with results is shown. So, I'm able to navigate in this window between various results. This what surprised me is that calculated P and T for particular calculations is considerably different from this what is shown in the TriQuick window. For example, calculated P and T seen in the results window is something like 6.3 kbar and 503 oC, while in the TriQuick window the red point is at 16 kbar and 503 oC. It is confusing. Furthermore, when I'm trying to inspect the reactions I obtain info that "reactions not calculated for this result". This is also strange as in the IR column there is 5. So, five independent reactions have been calculated. The same can be seen when I double click on this calculation in the Results window.
Definitely, something went wrong. However, I do not know how to solve this issue.
Best regards
Jacek
TC_Comb simple question
Dear Jacek,
Results (points and other figures) in TriQuick may be shifted to higher values on diagrams due to the general Windows system setting: scaling of screen contents (fonts etc.) to values other than 100%. Windows 10 developers have changed screen scaling technology while TriQuick uses old plotting component (which can not be updated). This is the first thing that comes to my mind. So, scaling = 100% is required in Windows screen parameters.
Do you see any reaction curves on calculated diagrams? If not, open the "Settings" window, tab "Reactions" and check "Calculate" checkbox. Actually, TC_Comb may run Thermocalc in two different modes consequently: "avPT" (code=2) and "Calculation of all reactions between endmembers" (code=3). Calculation of reaction curves takes a lot of time and not required in many cases, that is why it can be suppressed via this checkbox. Inspection of reactions require calculated reaction curves.
TC_Comb simple question
Dear Dima,
OK. I changed the scaling of my monitor and the red point in the TriQuick window is where it should be :-) Additionally, I checked the Calculate checkbox in the Reactions tab. Now, the reactions are calculated and can be inspected. Perfect!
Many thanks for your assistance!!!
Best regards
Jacek
Problems with graphs
Hi Dmitry,
I just discovered TC_Comb and I am excited trying the software. In my opinion it simplifies a lot the task of working with Thermocalc, which is not a very user friendly tool and it is helping me to better understanding of Thermocalc. Thank you very much for your work!
So far I have managed to do some calculations and by use of the previous answers in this forum I have been able to solve some questions. However, I have encountered another problem.
I am running avePT together with TriQuick but obtained graphs don't match with obtained the PT values... Although at first it seems that the calculated values are not plotted in the graph, using the mouse wheel, I modify the graph and I manage to see them. However, the values of P and T in the graph are very different from values in the results table in TC_Comb. I have tried to adjust values of X and Y in TriQuick but I do not succeed....
What can I be doing wrong?
Thank you very much in advance.
Best regards
Problems with graphs
Dear Sonya,
Thanks for the kind words about TC_Comb!
I recently encountered a similar issue with one of my users. This was caused by a scale setting in the display properties of newer versions of Windows: if the scale is not 100%, the old charting component used in the TriQuick program does not display plots correctly (with shifted coordinates). Unfortunately I can't fix it now, so the only solution is to set the scale to 100% in the screen properties. Hope this helps!
Sincerely,
Dmitriy
Now it works!! After change
Now it works!! After change the scale of the screen I can see the graphs properly.
Thank you very much!
Results Table Error
Dear Dmitry,
I am very excited to have discovered your program and I have started to experiment with my data to learn how to use TC_Comb. When the results window appears, it lists in the Notes column "Error calculating PT; Incomplete IR-set" and I am not sure how to remedy this. I am using Thermocalc version tc350-beta, the tc-ds55.txt database, and ax_2.exe. I have Grt, Bt, Ms and Pl as mineral entries into the program. I have tried different combinations of the end-members of these minerals, but I still get the same error message. I do not have data for all of the oxides for each mineral, so the entries in the columns appear as 0. Any help or advice you could provide would be really appreciated!
Thank you for your time!
Sincerely,
Taylor Morrell
Results Table Error
Dear Taylor,
THERMOCALC reports about incomplete set of (linearly) independent reactions (IR) when it cannot plot all IRs expected for this system within the specified PT-window. In this case, if total (expected) number of IRs is 3 or 4 but only 2 of them appear in the window, THERMOCALC rejects avPT calculations: the minimum IR number is 3 for this method. This is the reason of the message "Error calculating PT". It seems that your mineral compositions are too far from equilibrium: reactions are very scattered over the PT space. But try expanding the PT window as well: in many cases a very large window (say, 1-20 kbar and 100-1500°C) can help to find missing "runaway" reactions.
Regards,
Dmitry
AX2 and AX62 programs: new site and updates
NB! Tim Holland moved programs for calculation of activities AX2 and AX62 to an independent site: "AX : calculate activities of mineral endmembers from chemical analyses". The updated version (Oct 2022) fixed bugs in ternary feldspars and biotite.
"Error calculating PT; Incomplete IR-set"
Dear Dmitry,
Thanks for creating this very useful program, I am excited to do average PT calculations by using it. Unfortunately, I have continuously faced the same error "Error calculating PT; Incomplete IR-set", which has been mentioned by Taylor previously. To see what was going wrong, I tried to use the same mineral data from the teaching video TC Comb on Youtube as my input and followed the instructions in the video, but the error still occurred. The TC program I used is tc350-beta.exe, the AX program is AX_62.exe, and the database tc-ds62.txt. The P-T range for computation is 1-30 kbar and 300-1500 C. I have no idea how to overcome this obstacle, and I would greatly appreciate any help or advice you could offer, many thanks!
Best regard,
Hans
"Error calculating PT; Incomplete IR-set"
Dear Hans,
Is the version of TC actually 3.50-beta? As far as I know, the last version which supports old traditional avPT mode in TC_Comb is 3.45 (or, possibly, 3.47 - which may use slightly different keyboard input recorded in kbd-macros). TC_Comb with the newest TC versions should report about timeout due to inconsistent commands in avPT.kbd. In any case, you can select calculation mode in the TC_Comb window ("aver. PT" or "Reactions"), double-click on any line in the table with results and look at the actual TC output in a separate window, this can help to understand and localize the problem.
Hi Dmitry,
Hi Dmitry,
Thanks for responding so quickly. Using the TC version 3.45 from TC_AX_archive file, the program now works well!